
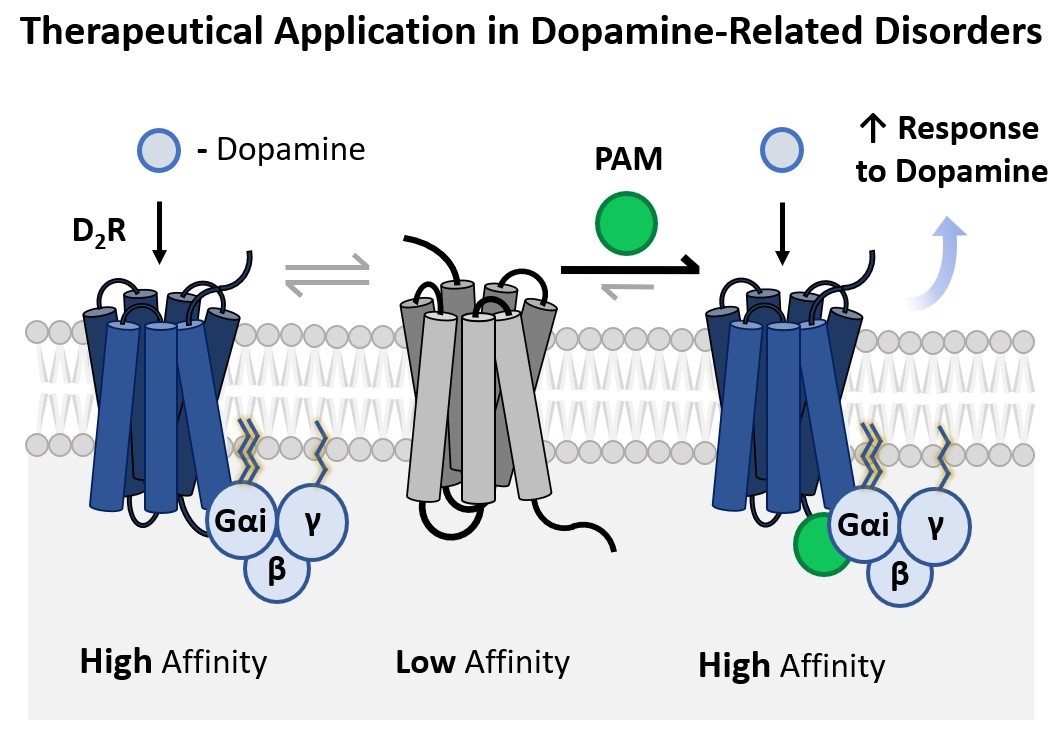
This invention concerns an improved synthetic methodology for the preparation of two cyclic aza-amino acid derivates and a new efficient protocol for the application of these compounds for the assembly of melanostatin-based azapeptides. The new azapeptides possess potent pharmacological activity as positive allosteric modulators (PAM) of the dopamine D₂ receptors at nanomolar concentrations, making them applicable in dopamine-related disorders, such as Parkinson's disease.
Melanostatin is a short endogeneous neuropeptide that displays potent PAM activity at the dopamine D₂ receptors, thus being of clinical interest as a therapeutical for dopamine-related disorders such as Parkinson's disease, as corroborated in clinical trials. However, its unfavorable pharmacokinetic properties such as low gastrointestinal absorption and reduced biochemical stability in neuronal tissues hinder its further development. Previous research has shown that the replacement of L-proline by D-proline has no significant impact on the pharmacological activity.
Moreover, since the metabolism of melanostatin occurs mainly upon cleavage of the prolyl-leucyl peptide bond, the replacement of this peptide bond with a semicarbazide moiety in melanostatin azapeptides is expected to increase biochemical resistance against protease activity without compromising the PAM activity. The incorporation of aza-amino acids in detriment to canonical amino acids is known to enhance pharmacological activity and selectivity as well as improve pharmacokinetic properties. Aza-proline is being employed as a proline surrogate in bioactive compounds as a chemical strategy to enhance potency and bioavailability.
- Cheap, straightforward, and high-yielding (tri)phosgene-free methodology for the preparation of stable aza-proline and aza-pipecolic acid carbazates, compatible with multi-gram scale for the assembly of azapeptides.
- Does not require anhydrous conditions for hydrazine cyclization, using NaOH as the base in the presence of phase-transfer agents.
- A more efficient, milder, and faster process for the application of aza-proline and aza-pipecolic acid carbazates for the preparation of azapeptides, operating at lower temperatures and offering higher yields.
- The methodology can be applied for the assembly of melanostatin azapeptides, with potential therapeutical application in the early stages of Parkinson's disease to delay the clinical use of levodopa therapy and/or to be used in co-administration to reduce the therapeutic doses of levodopa.
This invention provides a new method to develop intermediates for azapeptides synthesis, as well as a prospective active pharmaceutical ingredient (API) suitable for the treatment of dopamine-related diseases.




